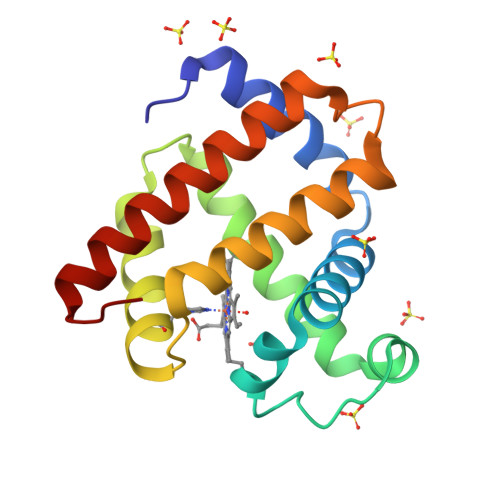
Goniometer-based femtosecond crystallography with X-ray free electron lasers.
Cohen, A.E., Soltis, S.M., Gonzalez, A., Aguila, L., Alonso-Mori, R., Barnes, C.O., Baxter, E.L., Brehmer, W., Brewster, A.S., Brunger, A.T., Calero, G., Chang, J.F., Chollet, M., Ehrensberger, P., Eriksson, T.L., Feng, Y., Hattne, J., Hedman, B., Hollenbeck, M., Holton, J.M., Keable, S., Kobilka, B.K., Kovaleva, E.G., Kruse, A.C., Lemke, H.T., Lin, G., Lyubimov, A.Y., Manglik, A., Mathews, I.I., McPhillips, S.E., Nelson, S., Peters, J.W., Sauter, N.K., Smith, C.A., Song, J., Stevenson, H.P., Tsai, Y., Uervirojnangkoorn, M., Vinetsky, V., Wakatsuki, S., Weis, W.I., Zadvornyy, O.A., Zeldin, O.B., Zhu, D., Hodgson, K.O.(2014) Proc Natl Acad Sci U S A 111: 17122-17127
- PubMed: 25362050
- DOI: https://doi.org/10.1073/pnas.1418733111
- Primary Citation of Related Structures:
4PNJ - PubMed Abstract:
The emerging method of femtosecond crystallography (FX) may extend the diffraction resolution accessible from small radiation-sensitive crystals and provides a means to determine catalytically accurate structures of acutely radiation-sensitive metalloenzymes. Automated goniometer-based instrumentation developed for use at the Linac Coherent Light Source enabled efficient and flexible FX experiments to be performed on a variety of sample types. In the case of rod-shaped Cpl hydrogenase crystals, only five crystals and about 30 min of beam time were used to obtain the 125 still diffraction patterns used to produce a 1.6-Å resolution electron density map. For smaller crystals, high-density grids were used to increase sample throughput; 930 myoglobin crystals mounted at random orientation inside 32 grids were exposed, demonstrating the utility of this approach. Screening results from cryocooled crystals of β2-adrenoreceptor and an RNA polymerase II complex indicate the potential to extend the diffraction resolution obtainable from very radiation-sensitive samples beyond that possible with undulator-based synchrotron sources.
Organizational Affiliation:
Stanford Synchrotron Radiation Lightsource, HodgsonK@stanford.edu acohen@slac.stanford.edu.