



New Features
Announcement and FAQ: EDMAPS.rcsb.org Shutdown October 16
09/24 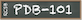
The EDMAPS.rcsb.org service for mtz files will be retired on October 16, 2024 (see previous announcement). This service has been providing RCSB PDB-calculated electron density map coefficients for X-ray structures in the PDB archive. In June, this service stopped providing DSN6 formatted maps for these structures.
Electron density map coefficients will instead be provided by wwPDB for all X-ray structures in the PDB archive. These map coefficients will be the same as those used in wwPDB Validation Reports. All validation data will be available from the public archive at https://files.wwpdb.org/pub/pdb/validation_reports/.
These validation data are also available from RCSB.org Structure Summary pages. Electron density map coefficients are available from the Download Files drop down menu (see 8WCG for an example).
Mol* at RCSB.org currently utilizes these electron density map coefficients in the Electron Density view.
Support for this transition is described below; additional questions can be sent to info@rcsb.org.
Using Validation Report Map Coefficient Files
wwPDB Validation Reports provide users with detailed reports that include an analysis of the quality of coordinate model and experimental data. The resulting analysis is available in PDF, XML and CIF formats. In addition, for X-ray crystallography structures, the coefficients used to produce electron density maps are provided for download. These coefficient files contain the data that can be used to produce electron density maps. All validation data will be available from the public archive at https://files.wwpdb.org/pub/pdb/validation_reports/.
At RCSB.org, these electron density map data can be visualized in the “Structure” tab of a structure summary page by pressing on the “density” option from the right hand menu and then pressing the enable button. Mol* at RCSB.org currently utilizes these electron density map coefficients in the Electron Density view accessible from under the molecular image on the Structure Summary page or by enabling the “density” option from the right-hand menu in Mol*.
Downloading Structure Factor File or Validation Map Coefficients
Assuming a UNIX based operating system, one can download the validation produced electron density map coefficient files. It can also be done manually, but this simple shell script will download the map coefficients for 2Fo-Fc and Fo-Fc maps.
#!/bin/sh
pdbid=$1
hash=${pdbid:1:2}
echo $pdbid $hash
base="https://files.rcsb.org/pub/pdb/validation_reports"
vdir=$base/$hash/$pdbid wget $vdir/${pdbid}_validation_2fo-fc_map_coef.cif.gz
wget $vdir/${pdbid}_validation_fo-fc_map_coef.cif.gz
if [ -e ${pdbid}_validation_2fo-fc_map_coef.cif.gz ]; then gunzip ${pdbid}_validation_2fo-fc_map_coef.cif.gz
fi
if [ -e ${pdbid}_validation_fo-fc_map_coef.cif.gz ]; then gunzip ${pdbid}_validation_fo-fc_map_coef.cif.gz
fi
Converting Experimental Data to MTZ File Format
There are several options to convert to a MTZ formatted file for use in Phenix or other CCP4 programs. Two of the easiest are `cif2mtz` and `gemmi`. Both are distributed with CCP4, but the gemmi program is available for standalone use and may be installed with a local Python installation.
For instance:
cif2mtz hklin xxxx_validation_2fo-fc_map_coef.cif hklout 2fo.mtz << EOF
EOF
or
gemmi cif2mtz xxxx_validation_2fo-fc_map_coef.cif 2fo.mtz
With gemmi, one can combine multiple coefficient files at the same time:
gemmi cif2mtz xxxx_validation_2fo-fc_map_coef.cif --add xxx_validation_fo-fc_map_coef.cif comb.mtz
As mentioned above, COOT can utilize such a combined file to visualize the electron density maps.
Using MTZ Map Coefficient Files to Produce CCP4 Maps
There are several ways to convert the map coefficient files to a CCP4 map. Only two are described here. Such map files can be used in Chimera, Pymol and other visualization software.
With Gemmi
gemmi sf2map 2fo.mtz 2fo.map
Or more directly: gemmi sf2map xxxx_validation_2fo-fc_map_coef.cif 2fo.map
The other common means to convert the file is with the CCP4 program FFT. This is what is used by RCSB.org.
fft HKLIN 2fo.mtz MAPOUT 2fo.map << EOF
LABIN F1=FWT PHI=PHWT
END
EOF
For an fo-fc map, use
F1=DELFWT SIGI=SIGFP PHI=PHDELWT
References
- COOT: Emsley P, Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D60, 2126-2132.
- Pymol: Schrödinger, L., & DeLano, W. (2020). PyMOL. Retrieved from http://www.pymol.org/pymol
- CCP4 Suite (for FFT): J. Agirre et al. Acta. Cryst. D79, 449-461 (2023) “The CCP4 suite: integrative software for macromolecular crystallography” [doi:10.1107/S2059798323003595].

pdbid=$1
hash=${pdbid:1:2}
echo $pdbid $hash
base="https://files.rcsb.org/pub/pdb/validation_reports"
vdir=$base/$hash/$pdbid wget $vdir/${pdbid}_validation_2fo-fc_map_coef.cif.gz
wget $vdir/${pdbid}_validation_fo-fc_map_coef.cif.gz
if [ -e ${pdbid}_validation_2fo-fc_map_coef.cif.gz ]; then gunzip ${pdbid}_validation_2fo-fc_map_coef.cif.gz
fi
if [ -e ${pdbid}_validation_fo-fc_map_coef.cif.gz ]; then gunzip ${pdbid}_validation_fo-fc_map_coef.cif.gz
fi
EOF
LABIN F1=FWT PHI=PHWT
END
EOF
