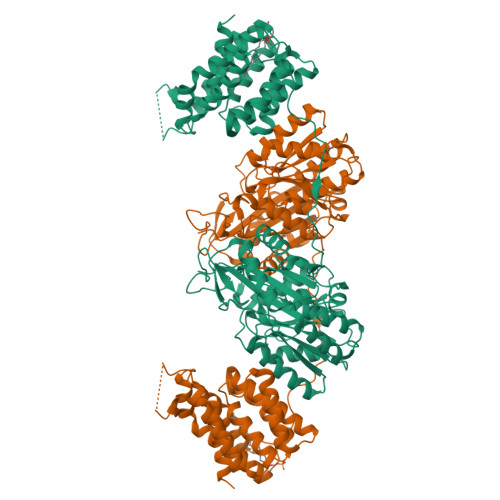
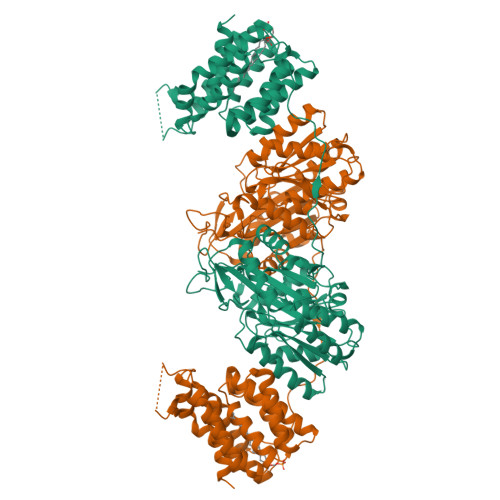
Crystal Structure of the Citrobacter Freundii Dihydroxyacetone Kinase Reveals an Eight-Stranded Alpha-Helical Barrel ATP-Binding Domain
Siebold, C., Arnold, I., Garcia-Alles, L.F., Baumann, U., Erni, B.(2003) J Biological Chem 278: 48236
- PubMed: 12966101
- DOI: https://doi.org/10.1074/jbc.M305942200
- Primary Citation of Related Structures:
1UN8, 1UN9 - PubMed Abstract:
Dihydroxyacetone kinases are a sequence-conserved family of enzymes, which utilize two different phosphoryldonors, ATP in animals, plants and some bacteria, and a multiphosphoprotein of the phosphoenolpyruvate carbohydrate phosphotransferase system in bacteria. Here we report the 2.5-A crystal structure of the homodimeric Citrobacter freundii dihydroxyacetone kinase complex with an ATP analogue and dihydroxyacetone. The N-terminal domain consists of two alpha/beta-folds with a molecule of dihydroxyacetone covalently bound in hemiaminal linkage to the N epsilon 2 of His-220. The C-terminal domain consists of a regular eight-helix alpha-barrel. The eight helices form a deep pocket, which includes a tightly bound phospholipid. Only the lipid headgroup protrudes from the surface. The nucleotide is bound on the top of the barrel across from the entrance to the lipid pocket. The phosphate groups are coordinated by two Mg2+ ions to gamma-carboxyl groups of aspartyl residues. The ATP binding site does not contain positively charged or aromatic groups. Paralogues of dihydroxyacetone kinase also occur in association with transcription regulators and proteins of unknown function pointing to biological roles beyond triose metabolism.
Organizational Affiliation:
Departement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland.