



The C-Ring Ion-Binding Site of the ATP Synthase from Bacillus Pseudofirmus of4 is Adapted to Alkaliphilic Lifestyle.
Preiss, L., Langer, J.D., Hicks, D.B., Liu, J., Yildiz, O., Krulwich, T.A., Meier, T.(2014) Mol Microbiol 92: 973
- PubMed: 24707994
- DOI: https://doi.org/10.1111/mmi.12605
- Primary Citation of Related Structures:
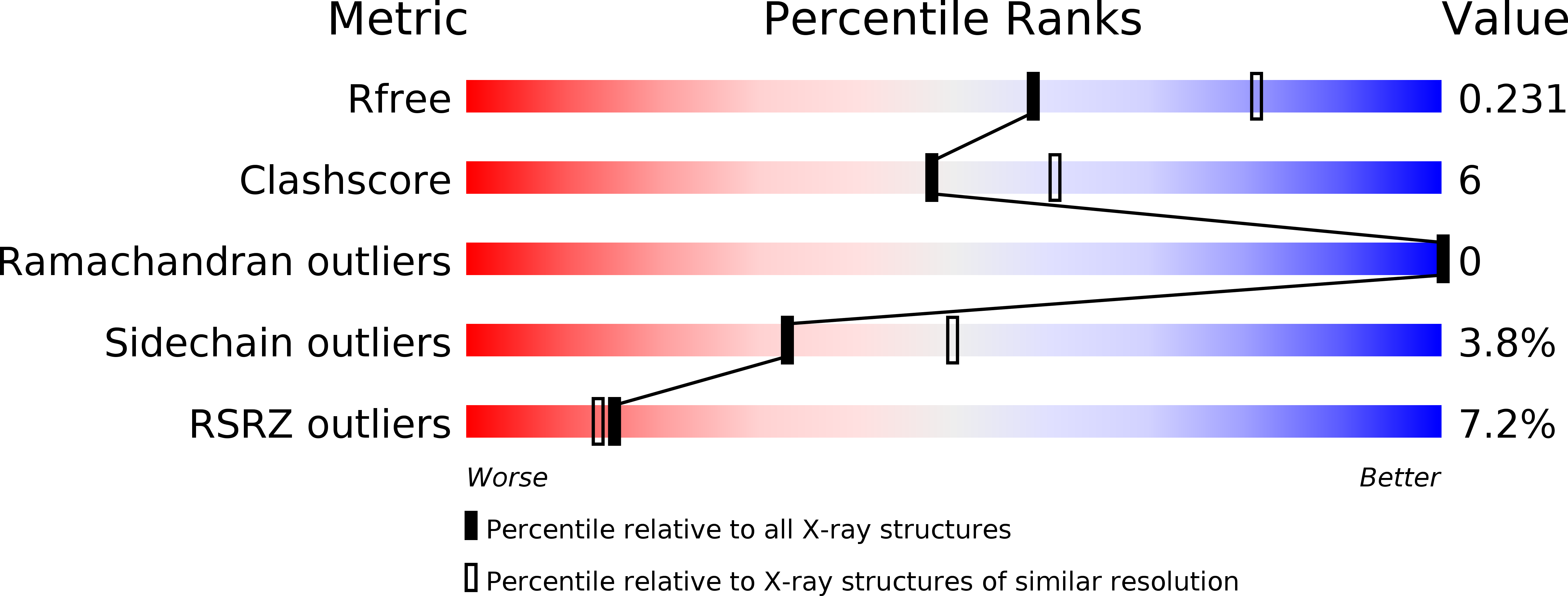
4CBJ, 4CBK - PubMed Abstract:
In the c-ring rotor of ATP synthases ions are shuttled across the membrane during ATP synthesis by a unique rotary mechanism. We investigated characteristics of the c-ring from the alkaliphile Bacillus pseudofirmus OF4 with respect to evolutionary adaptations to operate with protons at high environmental pH. The X-ray structures of the wild-type c13 ring at pH 9.0 and a 'neutralophile-like' mutant (P51A) at pH 4.4, at 2.4 and 2.8 Å resolution, respectively, reveal a dependency of the conformation and protonation state of the proton-binding glutamate (E(54) ) on environmental hydrophobicity. Faster labelling kinetics with the inhibitor dicyclohexylcarbodiimide (DCCD) demonstrate a greater flexibility of E(54) in the mutant due to reduced water occupancy within the H(+) binding site. A second 'neutralophile-like' mutant (V21N) shows reduced growth at high pH, which is explained by restricted conformational freedom of the mutant's E(54) carboxylate. The study directly connects subtle structural adaptations of the c-ring ion binding site to in vivo effects of alkaliphile cell physiology.
Organizational Affiliation:
Department of Structural Biology, Max Planck Institute of Biophysics, Max-von-Laue-Str. 3, 60438, Frankfurt am Main, Germany.









