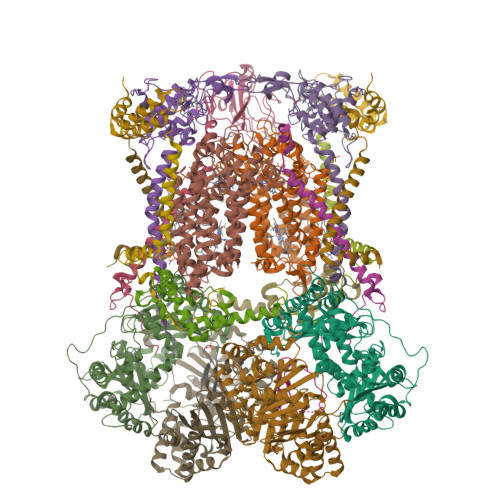
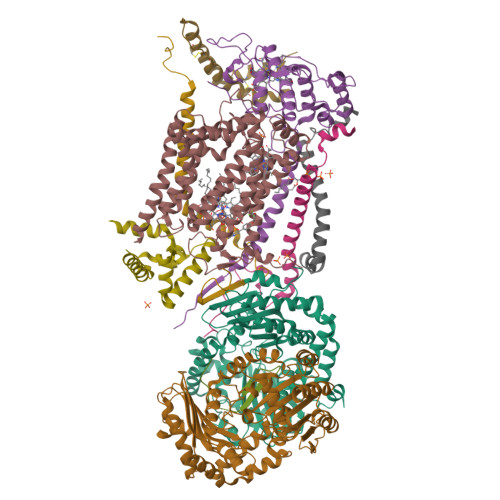
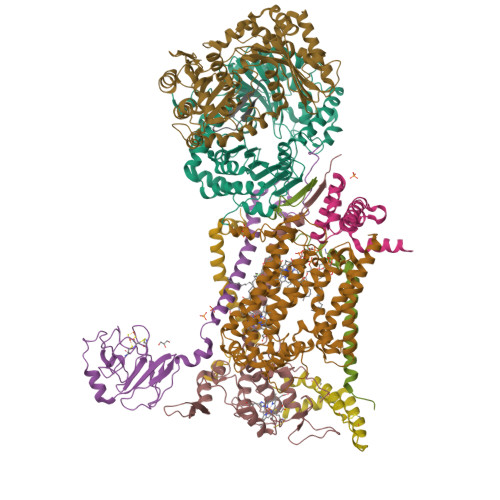
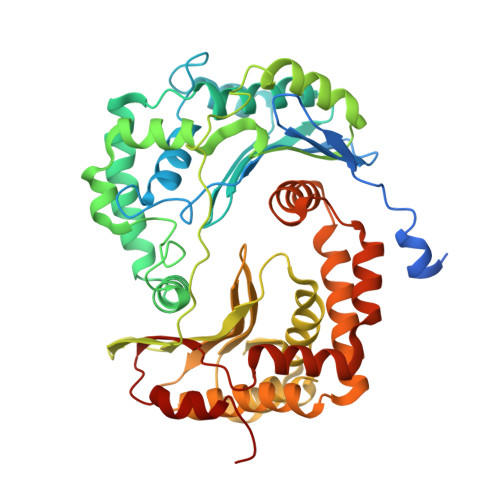
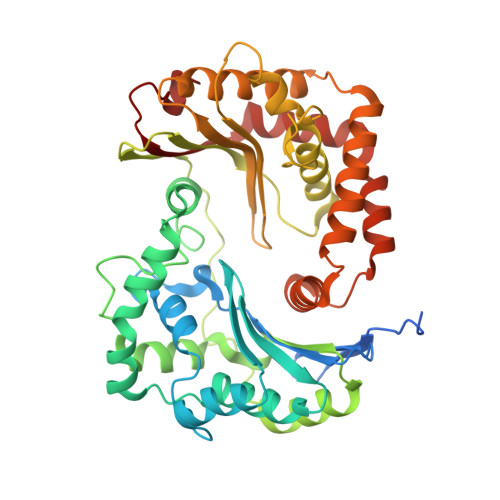
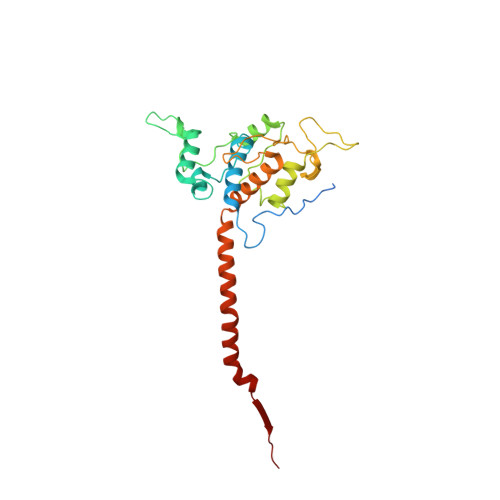
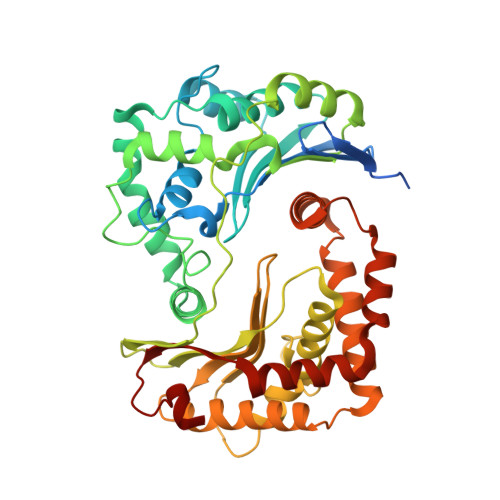
Antimalarial 4(1H)-Pyridones Bind to the Qi Site of Cytochrome Bc1.
Capper, M.J., O'Neill, P.M., Fisher, N., Strange, R.W., Moss, D., Ward, S.A., Berry, N.G., Lawrenson, A.S., Hasnain, S.S., Biagini, G.A., Antonyuk, S.V.(2015) Proc Natl Acad Sci U S A 112: 755
- PubMed: 25564664
- DOI: https://doi.org/10.1073/pnas.1416611112
- Primary Citation of Related Structures:
4D6T, 4D6U - PubMed Abstract:
Cytochrome bc1 is a proven drug target in the prevention and treatment of malaria. The rise in drug-resistant strains of Plasmodium falciparum, the organism responsible for malaria, has generated a global effort in designing new classes of drugs. Much of the design/redesign work on overcoming this resistance has been focused on compounds that are presumed to bind the Q(o) site (one of two potential binding sites within cytochrome bc1 using the known crystal structure of this large membrane-bound macromolecular complex via in silico modeling. Cocrystallization of the cytochrome bc1 complex with the 4(1H)-pyridone class of inhibitors, GSK932121 and GW844520, that have been shown to be potent antimalarial agents in vivo, revealed that these inhibitors do not bind at the Q(o) site but bind at the Q(i )site. The discovery that these compounds bind at the Q(i) site may provide a molecular explanation for the cardiotoxicity and eventual failure of GSK932121 in phase-1 clinical trial and highlight the need for direct experimental observation of a compound bound to a target site before chemical optimization and development for clinical trials. The binding of the 4(1H)-pyridone class of inhibitors to Q(i) also explains the ability of this class to overcome parasite Q(o)-based atovaquone resistance and provides critical structural information for future design of new selective compounds with improved safety profiles.
Organizational Affiliation:
Molecular Biophysics Group, Institute of Integrative Biology, Faculty of Health and Life Sciences, University of Liverpool, Liverpool L69 7ZB, United Kingdom;