Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors.
Vogerl, K., Ong, N., Senger, J., Herp, D., Schmidtkunz, K., Marek, M., Muller, M., Bartel, K., Shaik, T.B., Porter, N.J., Robaa, D., Christianson, D.W., Romier, C., Sippl, W., Jung, M., Bracher, F.(2019) J Med Chem 62: 1138-1166
- PubMed: 30645113
- DOI: https://doi.org/10.1021/acs.jmedchem.8b01090
- Primary Citation of Related Structures:
5W5K - PubMed Abstract:
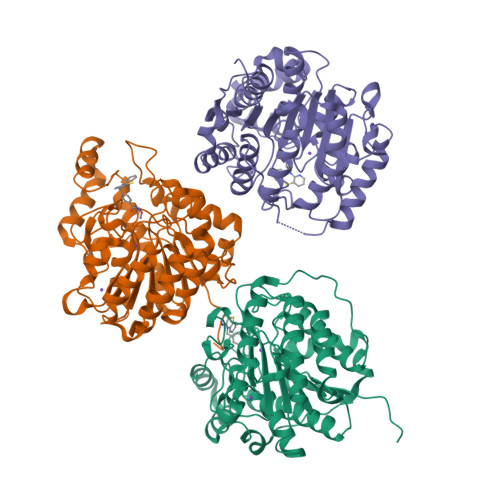
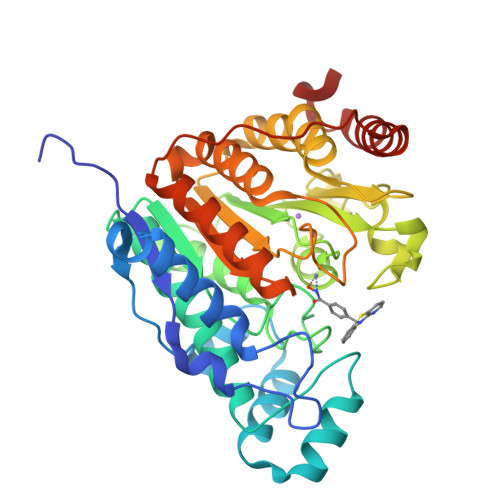
The phenothiazine system was identified as a favorable cap group for potent and selective histone deacetylase 6 (HDAC6) inhibitors. Here, we report the preparation and systematic variation of phenothiazines and their analogues containing a benzhydroxamic acid moiety as the zinc-binding group. We evaluated their ability to selectively inhibit HDAC6 by a recombinant HDAC enzyme assay, by determining the protein acetylation levels in cells by western blotting (tubulin vs histone acetylation), and by assessing their effects on various cancer cell lines. Structure-activity relationship studies revealed that incorporation of a nitrogen atom into the phenothiazine framework results in increased potency and selectivity for HDAC6 (more than 500-fold selectivity relative to the inhibition of HDAC1, HDAC4, and HDAC8), as rationalized by molecular modeling and docking studies. The binding mode was confirmed by co-crystallization of the potent azaphenothiazine inhibitor with catalytic domain 2 from Danio rerio HDAC6.
Organizational Affiliation:
Department of Pharmacy-Center for Drug Research , Ludwig-Maximilians University Munich , Butenandtstr. 5-13 , 81377 Munich , Germany.