



RCSB PDB Help
Search and Browse > Advanced Search
Sequence Similarity Search
Introduction
The order of amino acids in a protein or the protein sequence is thought to hold the key for protein folding, i.e., each protein sequence folds into a unique shape to perform its function(s). While this relationship between sequence and structure holds true in most of the cases, some folded proteins may adopt multiple conformations corresponding to different functional states and a few protein sequences can fold into entirely different shapes (e.g., prion proteins). Sequence comparison is the most commonly used strategy for identifying homology between proteins and/or protein domains.
What is Sequence Similarity Search?
The Sequence Similarity Search option allows you to query the PDB archive using the amino acid sequence of a protein. Although the most common method for sequence similarity searches is Basic Local Alignment Search Tool (BLAST, Altschul et al., 1990), the Sequence Similarity Search option available from RCSB.org uses the MMseqs2 software (Steinegger and Söding, 2017) to find similar protein and nucleic acid sequences. The MMseqs2 tool is similar to BLAST, but achieves better performance at comparable levels of sensitivity. The search is dependent on a user-defined "sequence identity cutoff". During the search, each structure in the archive is aligned with the target structure, and the number of identical amino acids is tallied. If this number is greater than the cutoff, the structure is returned by the search.
Why run a Sequence Similarity Search?
Sequence similarity searches are widely used to find similar proteins in the archive and identify conserved domains in them.
Aligning a query sequence with other sequences in a database or aligning a group of similar sequences and statistically assessing how well they match can help identify homology between proteins. This can be used to transfer information and develop hypotheses about the protein’s interactions and functions.
For example, a search of sequences similar to that of whale myoglobin using a high cutoff value for sequence identity (~90%) will return a list of additional structure determinations of the particular protein (i.e., the whale myoglobin). Using a lower cutoff value (~30%) for the same search will return a longer list that includes myoglobin structures from other organisms and other proteins with similar structure, e.g., cytoglobin.
Documentation
There are a few different options that can be combined to run a Sequence Similarity Search. These options are being listed here under 3 different sections:
- Query - this will describe the options you have to inputting your query
- Search - this will describe the search parameters and filters that can be applied.
- Results - this will describe options available for what you wish to see in the results page.
Query Options
A Sequence Similarity Search query must be at least 25 residues long and be submitted in FASTA format (amino acid one letter code). For shorter sequences try the Sequence Motif Search. The sequence similarity search query can be submitted in two ways - either using the Advanced Search interface or from the Structure Summary Page.
Query using the Top Bar Search box
Pasting the one-letter code of a protein, DNA, or RNA sequence in the top bar search box is recognized as a sequence similarity search request (Figure 1A). This sequence is automatically transferred to the Advanced Search Query Builder’s Sequence Similarity search and a search is performed for experimental structures and Computed Structure Models (CSMs) with the same or similar sequence (Figure 1B).
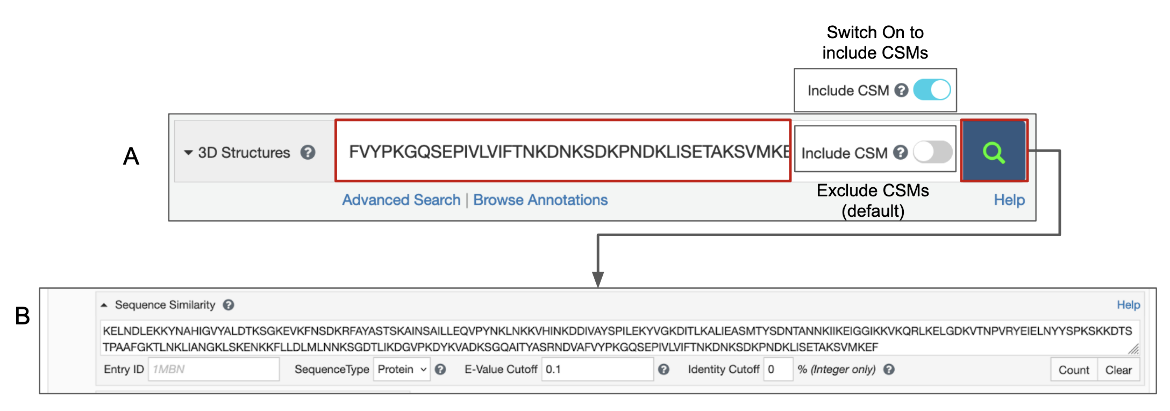
|
| Figure 1: Options for sequence similarity search from the Top bar search box - A. paste FASTA sequence; B. the query sequence is transfered to the Sequence Similarity search options in the Advanced Search Query Builder to run the search. Note: Turn on the toggle switch to Include CSMs. |
Query using the 'Advanced Search' panel
The sequence similarity search options available from the “Advanced Search” panel are as follows:
The query can be submitted using the PDB ID. This is useful to find all sequences that are similar to the sequence from a specified chain.
- Type in a structure ID (e.g., PDB ID or RCSB.org assigned CSM ID) into the appropriate box. Once you enter a valid structure ID the tool presents a list of macromolecular sequences with chain IDs in a pop-up box. Note that the sequences of all unique polymer entities are presented here (Figure 2A).
- Select a Chain ID and sequence of interest from the pull-down menu. This selected sequence will be pasted into the Sequence box above the PDB ID box (Figure 2B)
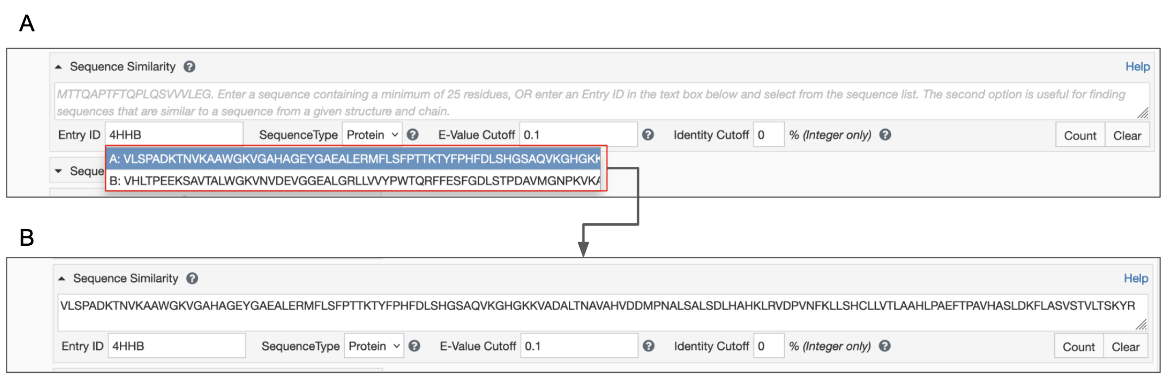
|
| Figure 2: Options for sequence similarity search from the Advanced Search Query Builder - A. input structure ID and select polymer chain of interest; B. the selected polymer chain sequence is populated in the Sequence similarity search box. |
The query can also be submitted as a sequence. This is useful when you have a FASTA format sequence on hand (either from a publication or a bioinformatics resource) to pasted inthe box provided (Figure 2B).
- Paste the sequence in one-letter code format in the Sequence text box (See Panel B in figure above). Be sure to remove any other information that might be at the top of the pasted sequence (e.g., FASTA headers).
Query from the Structure Summary page
Each structure in the PDB has a dedicated Structure Summary page that displays information about the entities in that entry. To search for polymer entities with sequences similar to that of a specific polymer entity click on the “Sequence” link above the details listed for the macromolecular entity and choose the desired sequence identity level from the drop-down (Figure 3). The search is run with the default E-value cutoff (0.1). This and other search options are explained further in the following section.
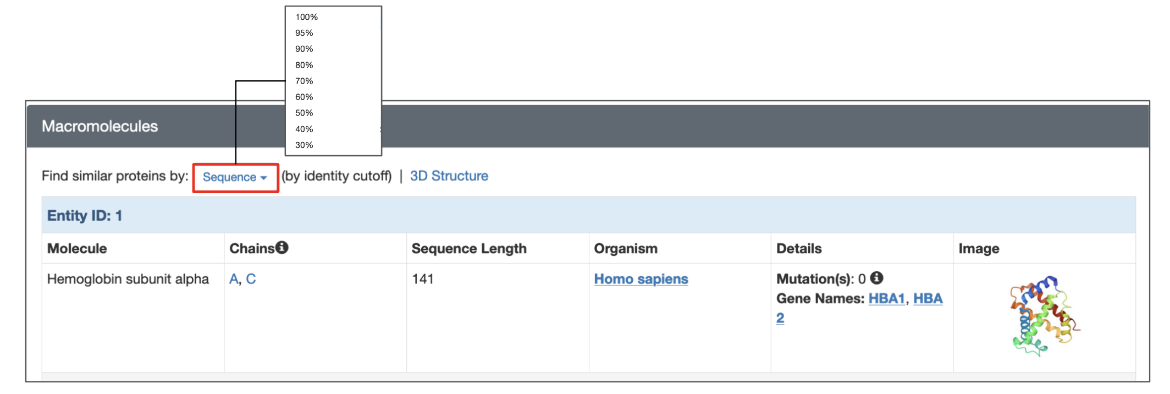
|
| Figure 3: Options for launching a sequence similarity search from the structure summary page of a specific 3D structure. |
Search Options
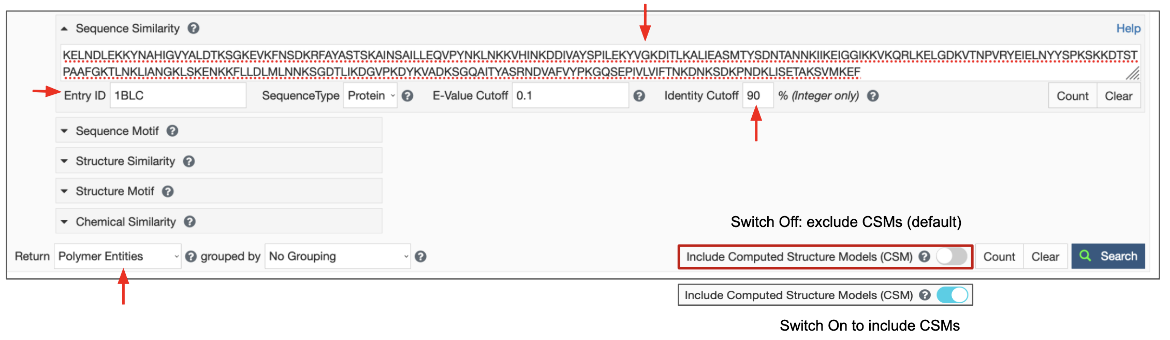
Once the Query sequence has been provided, the following parameters can be specified to launch the search (Figure 4). If they are not specified, reasonable defaults will be used.
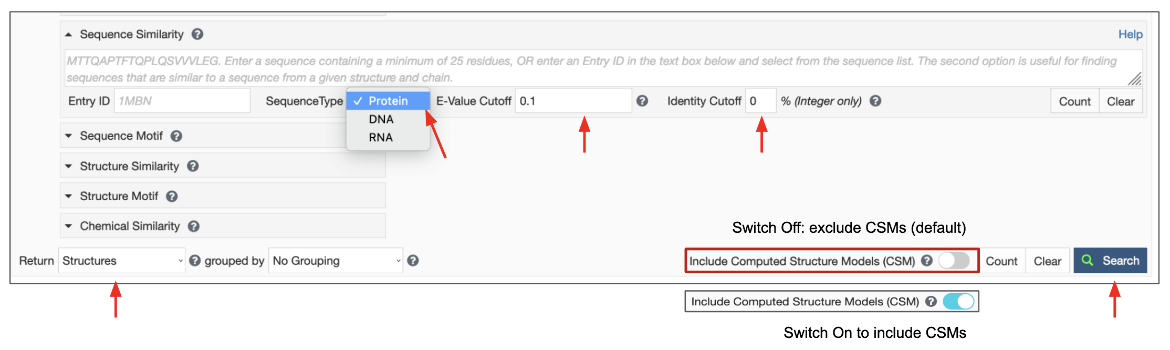
|
| Figure 4: Sequence Similarity Search parameters. |
- Search Target: There are three possible target databases for the sequence similarity search present in the PDB and in CSMs available from RCSB.org - (1) all current protein sequences, (2) all current DNA sequences and (3) all current RNA sequences.
- E-value Cutoff: The E-value or Expect value is a parameter that describes the probability that the sequences matched by chance. The lower the E-value, or the closer it is to zero, the more "significant" the match is. Sequences that have an E-value higher than the set cutoff are filtered out of the results. The default E-value for Sequence Similarity Search is set at 0.1 and is usually indicated in scientific notation, (e.g., 1e-10). You can change this to a smaller or larger number to meet the needs of your search. Since the scoring takes chain length into consideration shorter sequences can have identical matches with high E-Value.
- Identity Cutoff defines the extent of match between two sequences. The cutoff value is measured as a percentage, with a value between 0 and 100%. The default value is set to zero so all matched sequences are listed in the results. Select a level of identity for the sequence similarity search as needed. Matched sequences that have sequence identity lower than this cutoff are filtered out of the results.
Since the query for this search is a sequence, the results are expected to be sequences of polymer entities. Select the “Polymer Entities” option for the Display Results as pull-down list, use the default option to exclude CSMs or turn on the toggle switch to include CSMs, and then launch the search by clicking on the Search button.
Results
At the top of the search results page, several options are available for displaying, reporting, sorting, and downloading all or selected entities, entries, and assemblies that match the query.
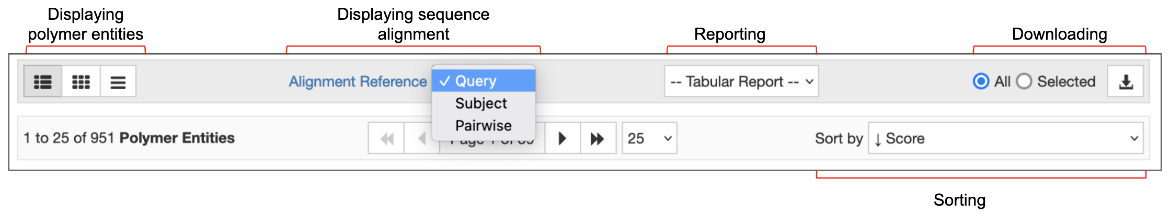
|
| Figure 5: Options to display, report, sort, and download search results. |
The default option for displaying the results is the Summary view. This includes a snapshot of the matched polymer entity, entry, or assembly, and lists the structure title, authors, brief information about the contents (polymer name, ligands), source organism and a few other details. Options are available to display the results in a Compact view, or as an image Gallery of the matched polymers, entries, or assemblies.
The Tabular Report options allow you to create detailed reports about the matched entries, entities, or assemblies. Using the sorting options, you can order the results by score, date of release, etc., while the download options allow users to download all or selected entries, entities, and assemblies.
The “Alignment Reference” options are unique to the sequence similarity search results. These options allow you to change the interactive graphical summaries of the sequence alignment. Learn more about these options and the color scheme used for alignment here.
Polymer entities that match the query and search parameters are listed on the Results page using the default “Alignment Reference” option (Figure 6).
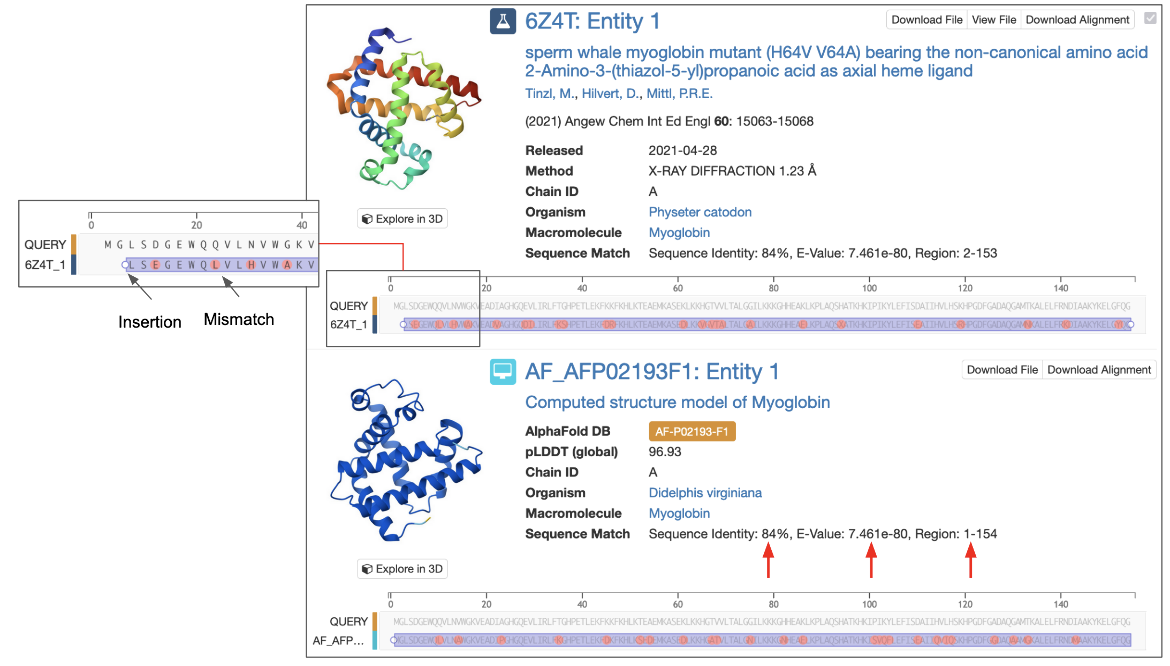
|
| Figure 6: Part of a sequence similarity search results page showing an experimental structure and a CSM that matched the query. The inset shows a zoomed in view of the sequence alignment including an insertion and several mismatches. Sequence similarity measures are highlighted with red arrows. |
The following values are presented to qualify the extent of the match:
- Sequence Identity is the ratio of the number of identical amino acids between the 2 aligned sequences over the aligned length, expressed as a percentage.
- E-Value provides a measure for whether the observed sequence identity was a chance match or if it has any evolutionary significance. This is reported as a number close to zero. The lower the E-Value, or the closer it is to zero, the more "significant" the match is. An E-Value of a significant match is often expressed in scientific notation, where the higher the exponent’s power the more significant the match (i.e., 1e-80 is a more significant match than 1e-30). Note that short query sequences may be seen in many other protein sequences by chance so these matches may have a high E-Value.
- Region specifies the amino acid residue numbers of the part of the protein that was considered in the sequence comparison.
You can explore the sequence match using the interactive alignment displayed. Zoom into the sequence and move the sequence left or right to examine sequence identities, mismatches, insertions, deletions or missing residues etc.
You can also download and view the matched files and/or the alignments.
Limitations of Sequence Similarity Search
Sequence similarity searches with short query sequences can match many sequences and provide false evolutionary relationships. This should be carefully examined and considered when making any conclusions about the matched sequences. For short sequences, the Sequence motif search may be more suitable.
Examples
1. Search for sequences that are 90% similar to the beta-lactamase in the PDB entry 1blc using the Advanced Search interface.
- Launch this search from the Advanced Search interface for PDB ID 1blc, Chain ID A.
- Provide the Identity Cutoff as 90 and select the Display results options from the pulldown menu to be Polymer Entities, and launch the search
|
| Figure 7: Setting up the sequence similarity search for the sequence in Chain A of PDB entry 1blc, at 90% sequence identity. Turn on the toggle switch to include CSMs. |
- The search results include several other beta lactamase entities but all from the same organism, Staphylococcus aureus.
2. Search for sequences that are 30% similar to the beta-lactamase in the PDB entry 1blc from the Structure Summary page (of PDB entry 1blc)
- Launch this search from the Structure Summary page for the PDB entry 1blc, Macromolecule Entity 1 (BETA-LACTAMASE).
- Click on the Sequence button and select the 30% option in the pulldown menu.
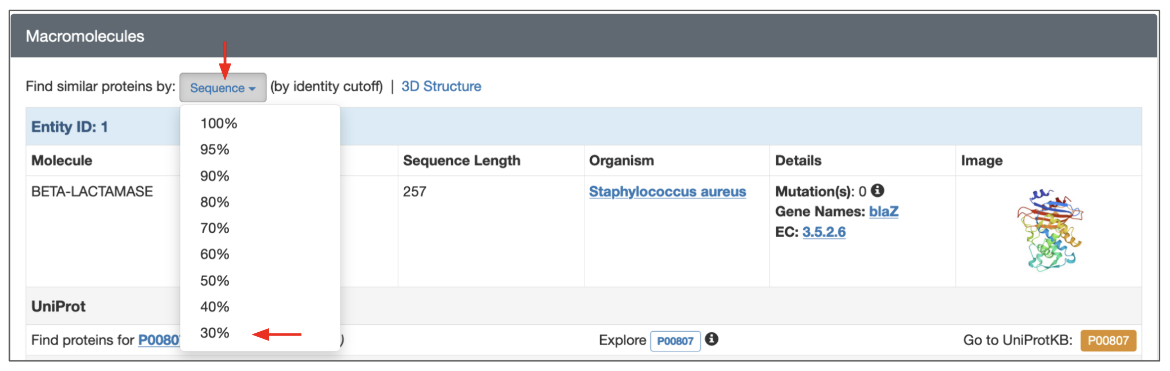
|
| Figure 8: Launching a sequence similarity search from the Structure Summary Page for PDB entry 1blc. |
- The search results include many other beta-lactamase structures from different organisms and specificities.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of molecular biology, 215(3), 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Steinegger, M., Söding, J. (2017). MMseqs2 enables sensitive protein sequence similarity searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028. https://doi.org/10.1038/nbt.3988


